|










:format(webp):quality(80)/https://romaniatv.net/wp-content/uploads/2014/08/520364_dinu_publimedia_cosmin_motei_56904200.jpg)











| Fluxul de Stiri dupa Data Aparitiei |
|
|
Comentarii Adauga Comentariu _ Obstacole de reglementare pentru actualizarea punctelor de întrerupere pentru dispozitivele de testare a sensibilității antimicrobiene: Ce trebuie să știți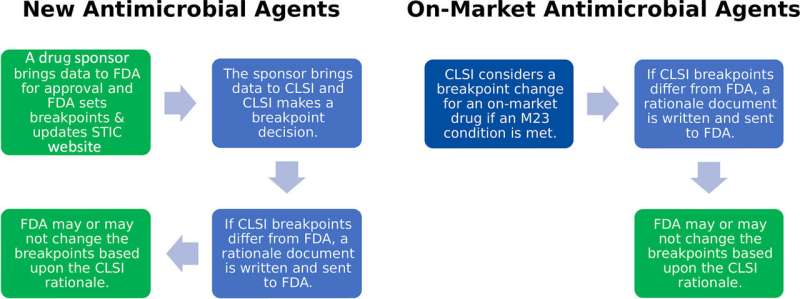 _ Obstacole de reglementare pentru actualizare Puncte de întrerupere pentru dispozitivele de testare a sensibilității antimicrobiene: Ce trebuie să știțiMediul de reglementare unic pentru eliminarea și utilizarea sistemelor de testare a sensibilității antimicrobiene (AST) este complex în S.U.A. În timp ce membrii comunității microbiologiei medicale recunosc importanța actualizarea punctelor de întrerupere pentru sănătatea clinică și publică, există o lipsă de cunoștințe privind cerințele de reglementare pentru actualizările punctelor de întrerupere și modul în care aceste cerințe afectează procesele de actualizare ale laboratorului medical. Ce trebuie să știe profesioniștii din laboratorul medical despre Alimente și Procesul de eliminare a punctelor de întrerupere de la Drug Administration (FDA) și modul în care acesta afectează testarea acestora? Două ramuri ale FDA sunt implicate în eliminarea punctelor de întrerupere pentru dispozitivele AST. Centrul FDA pentru Evaluare și Cercetare a Medicamentului (CDER) reglementează antimicrobienele și punctele de întrerupere asociate acestora. În schimb, Centrul FDA pentru Dispozitive și Sănătate Radiologică reglementează dispozitivele AST, inclusiv implementarea punctelor de întrerupere pe baza punctelor de întrerupere recomandate de CDER la momentul înregistrării dispozitivului. Dispozitivele medicale sunt clasificate în 3 categorii și cerințele de reglementare cresc pe măsură ce clasificarea trece de la clasa 1-3. Aceste categorii se bazează în primul rând pe utilizarea prevăzută a dispozitivului, indicațiile de utilizare și riscul pentru pacient sau utilizator. Dispozitivele de testare a sensibilității antimicrobiene sunt considerate dispozitive de clasa 2. Pentru ca un nou dispozitiv de clasa 2 să primească autorizație sau pentru ca o modificare a dispozitivului existent să fie eliminată, un document numit 510(k) trebuie trimis la FDA pentru revizuire. Potrivit FDA, „un 510(k) este o prezentare înainte de comercializare făcută către FDA pentru a demonstra că dispozitivul care urmează să fie comercializat este la fel de sigur și eficient, adică echivalent în mod substanțial cu un dispozitiv comercializat legal.” În cazul punctelor de întrerupere, producătorul dispozitivului poate fi nevoit să trimită date sub forma unui 510(k) care să demonstreze performanța acceptabilă a medicamentului pe dispozitiv atunci când se utilizează puncte de întrerupere noi. Pentru a eficientiza și a accelera calea de actualizare a punctului de întrerupere, în 2019, FDA a creat un proces prin care producătorii puteau trimite un protocol de modificare a punctului de întrerupere cu transmiterea lor 510(k) pentru aprobare. Scopul acestui protocol a fost de a permite producătorii să descrie în mod proactiv acțiunile pe care le-ar întreprinde pentru a actualiza informațiile privind punctul de întrerupere din eticheta produsului și, dacă condițiile ar fi adecvate, i-ar împiedica să trimită o altă cerere de reglementare la FDA în momentul actualizării punctului de întrerupere. Deși aceasta a fost o modificare utilă, au existat încă limitări privind modul în care ar putea fi utilizat protocolul. În septembrie 2023, FDA a emis un nou document de orientare mai cuprinzător pentru producătorii de AST, care înlocuiește documentul de orientare publicat de agenției în 2009 și oferă o opțiune pentru accelerarea în continuare a actualizărilor punctelor de întrerupere, numită un plan de control al schimbărilor predeterminat (PCCP), anterior plan de control al punctelor de întrerupere. PCCP poate fi urmărit de producătorii care trimit un original 510(k). ) documentul de autorizare pentru dispozitivul lor (de exemplu, un dispozitiv nou care vine pe piață) și cei care actualizează punctele de întrerupere pe un dispozitiv (vechi) șters anterior. Odată ce PCCP este șters cu o trimitere 510(k), în în cazul actualizărilor viitoare ale punctelor de întrerupere și dacă sunt îndeplinite anumite criterii, eticheta poate fi actualizată fără o prezentare suplimentară 510(k) la FDA. Avantajul principal al acestui program este că producătorii de dispozitive AST noi și vechi pot reprocesa datele existente pentru a demonstra performanța acceptabilă cu noul punct de întrerupere, pot trimite actualizarea la FDA și își pot actualiza eticheta pentru utilizare pe sistem. Acest proces revizuit economisește o cantitate semnificativă de timp, deoarece nu este necesară o revizuire a datelor de către FDA. Este important de reținut că actualizarea punctului de întrerupere nu ar trebui să modifice în mod semnificativ performanța dispozitivului șters anterior și, pentru ca un PCCP să fie aplicabil, regulamentul de clasificare a dispozitivului, codul produsului, utilizarea prevăzută și clasificările tehnologice ar trebui să fie aceleași cu cele șterse inițial. În plus, actualizările punctelor de întrerupere sunt de obicei urmate de scrisori ale clienților, alte forme de comunicare de la producător care trebuie construite și actualizări de software. Având în vedere aceste considerații, profesioniștii din laboratorul medical se pot aștepta la o decalaj între publicarea noilor puncte de întrerupere și implementarea completă a punctelor de întrerupere în sistemul lor automatizat, chiar dacă procesul de eliminare este accelerat. În sfârșit, producătorii pot utiliza numai procesul PCCP pentru punctele de întrerupere publicat pe site-ul FDA STIC, astfel încât acest proces se aplică numai punctelor de întrerupere CLSI pe care FDA le-a recunoscut. Înainte de 2007, atât punctele de întrerupere ale FDA, cât și ale Clinical and Laboratory Standards Institute (CLSI) ar putea fi raportate de la un dispozitiv AST aprobat de FDA. În 2007, FDA a publicat un document de orientare special de clasa II special pentru sistemele AST, care impunea ca dispozitivele AST din SUA să raporteze numai punctele de întrerupere recunoscute de FDA, iar utilizarea punctelor de întrerupere CLSI a fost permisă numai pentru instrumentele cu autorizații în 2007 sau mai devreme. După o revizuire în 2009, documentul a precizat că rezultatele de la dispozitivele AST ar trebui raportate numai pentru speciile microbiene care aveau date de eficacitate in vivo enumerate în instrucțiunile de utilizare ale antimicrobienilor. Astăzi, aceasta reprezintă o dilemă pentru producătorii de dispozitive AST care caută autorizație pentru noi puncte de întrerupere pe sistemele lor și pentru laboratoarele medicale care speră să le actualizeze. Nu mai este permisă „echivalarea” vechilor puncte de întrerupere sau a afirmațiilor cu privire la organism. Aceasta înseamnă că producătorii de dispozitive pot primi autorizație numai pentru punctele de întrerupere recunoscute de FDA și pentru organisme revendicate în eticheta antimicrobiană originală. În plus, lista de organisme cu date in vivo în eticheta medicamentului (cunoscută ca „lista 1”) este de obicei limitată și adesea nu conține toate organismele relevante din punct de vedere clinic care pot fi tratate în cadrul clinic. Aceste reglementări au un impact semnificativ asupra laboratoarelor medicale din mai multe motive. De exemplu, multe clearance-uri au fost obținute înainte de 2007 pentru categorii mari de organisme, cum ar fi „bacili gram negativi semnificativi clinic”. Din acest motiv, efectuarea testelor de susceptibilitate (folosind dispozitivul eliminat) pe toți bacilii gram negativi semnificativi clinic a fost considerată pe etichetă. În conformitate cu noile reglementări FDA, multe afirmații privind organismele pot fi eliminate atunci când autorizarea pentru noi se urmărește punctele de întrerupere, ceea ce face ca raportarea oricărui organism care nu este listat în prospectul testului să fie în afara etichetei. Să parcurgem un exemplu de utilizare a antibioticului cefazolin. Punctele de întrerupere pentru cefazolină au fost reduse de CLSI în 2010, iar FDA-CDER este de acord cu aceste puncte de întrerupere pentru infecțiile sistemice (nu infecțiile necomplicate ale tractului urinar). Deoarece FDA recunoaște punctele de întrerupere CLSI, producătorii de dispozitive AST pot trimite date care demonstrează performanța acelor puncte de întrerupere și solicită autorizație pentru dispozitivul lor. Cu toate acestea, deoarece afirmațiile despre organism trebuie să reflecte acum ceea ce este în prospectul cu antibioticul cefazolin, categoria largă de „bacili gram negativi semnificativi clinic” nu se mai aplică. În cazul cefazolinei, singurul gram gram. -organismele negative enumerate în etichetă pentru infecții sistemice sunt Escherichia coli și Proteus mirabilis. Producătorii au voie să trimită date care susțin testarea organismelor care nu sunt „lista 1”, atâta timp cât aceste organisme nu cuprind mai mult de 5% din setul de date și sunt similare din punct de vedere biologic cu organismele din „lista 1” (de exemplu, sunt toate Enterobacterales) . Dacă un prospect va conține sau nu numai speciile enumerate pe eticheta medicamentului sau un grup mai mare, cum ar fi Enterobacterales, va depinde de datele transmise de producător. Indiferent, acest regulament poate limita speciile indicate pe etichetă, deoarece includerea unor grupuri mari cu organisme neetichetate (de exemplu, bacili gram negativi semnificativi clinic) nu mai este permisă. Dacă un laborator medical dorește să utilizeze noile puncte de întrerupere pe dispozitivul lor pentru alte organisme gram-negative, acest lucru este considerat off-label, iar laboratorul trebuie să efectueze o validare. Dacă producătorul a efectuat o validare internă pentru combinații suplimentare organism-medicament, aceste date pot fi aplicabile în țările care urmează ghidul ISO 20776-2 pentru determinarea performanței sistemelor AST. Acest lucru se datorează faptului că standardele de performanță ISO. evaluează acordul esențial și părtinirea și sunt independente de punctele de întrerupere, ceea ce permite implementarea imediată a noilor puncte de întrerupere. Cu toate acestea, acest lucru diferă în S.U.A., unde producătorii de dispozitive trebuie să respecte cu strictețe reglementările FDA. Actualizarea punctelor de întrerupere este esențială pentru îngrijirea adecvată a pacientului și sănătatea publică și ar trebui să aibă prioritate de către laboratoarele de microbiologie medicală. Deși acest proces poate fi o întreprindere semnificativă, mai multe resurse sunt disponibile pentru a ajuta laboratoarele și pentru a le ajuta să mențină punctele de întrerupere AST actualizate în toate metodologiile lor de testare. Într-o publicație recentă care rezumă conferința ASM 2022 Clin Micro Open, publicată în Jurnalul de Microbiologie Clinică, autorii observă o lipsă semnificativă de înțelegere cu privire la barierele și nevoile dintre grupurile de microbiologie din industrie, de reglementare, clinice și de sănătate publică. Publicația solicită un dialog de grup mai mare și o colaborare între sectoare pentru a aborda barierele legate de actualizarea punctelor de întrerupere și de implementare. Profesioniștii din laboratoarele medicale se pot afla efectuând numeroase validări off-label pentru a utiliza punctele de întrerupere actualizate pe dispozitivul lor sau pentru a valida organismele acum. considerate în afara etichetei după o autorizare recentă a punctului de întrerupere. Utilizatorii de dispozitive AST trebuie să contacteze producătorul cu orice întrebări despre autorizare sau actualizări ale punctelor de întrerupere pentru a înțelege ce poate necesita validare și planifica în consecință. În plus, este imperativ să colaborați cu colegii clinici pentru a prioritiza validările punctelor de întrerupere și a organismelor în funcție de nevoi, resurse și epidemiologia locală.
Linkul direct catre PetitieCitiți și cele mai căutate articole de pe Fluierul:
|
|
|
















































Comentarii:
Adauga Comentariu